
Estuarine Aquacultures at the Crossroads of Animal Production and Antibacterial Resistance: A Metagenomic Approach to the Resistome
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Sample Collection and Preparation
2.2. Genomic Analysis
2.3. Statistical Analysis
3. Results
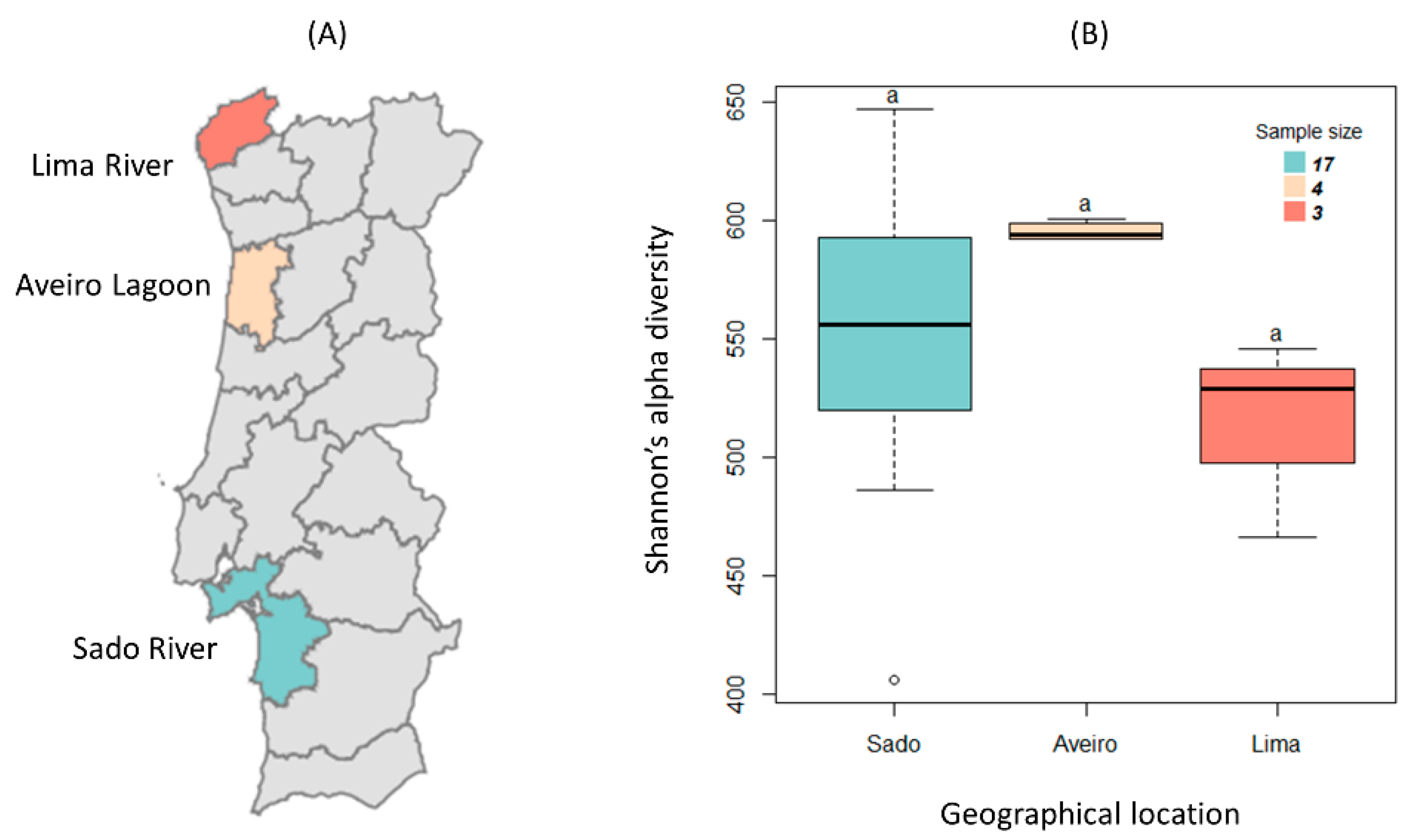
3.1. Sample Characterization
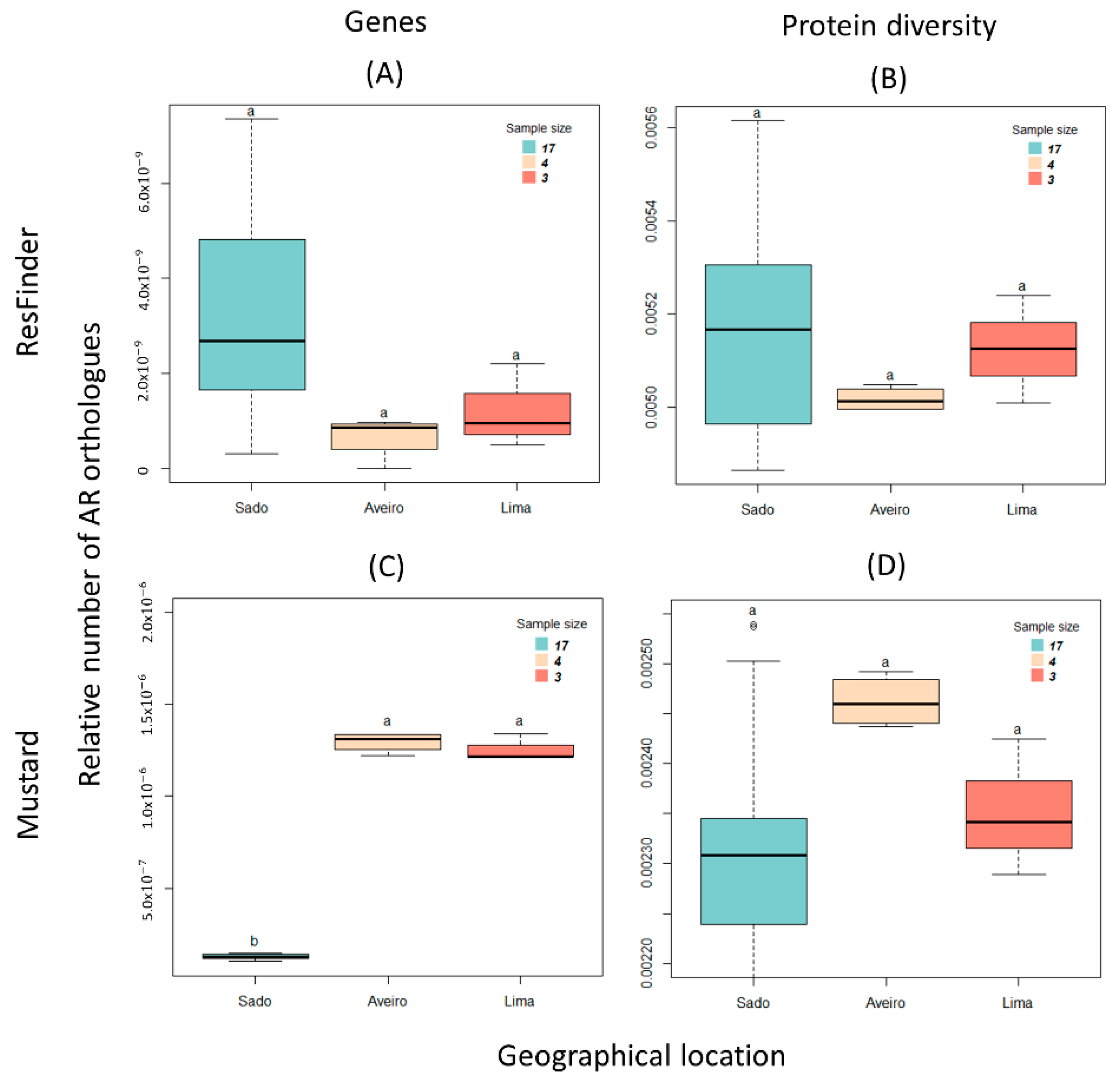
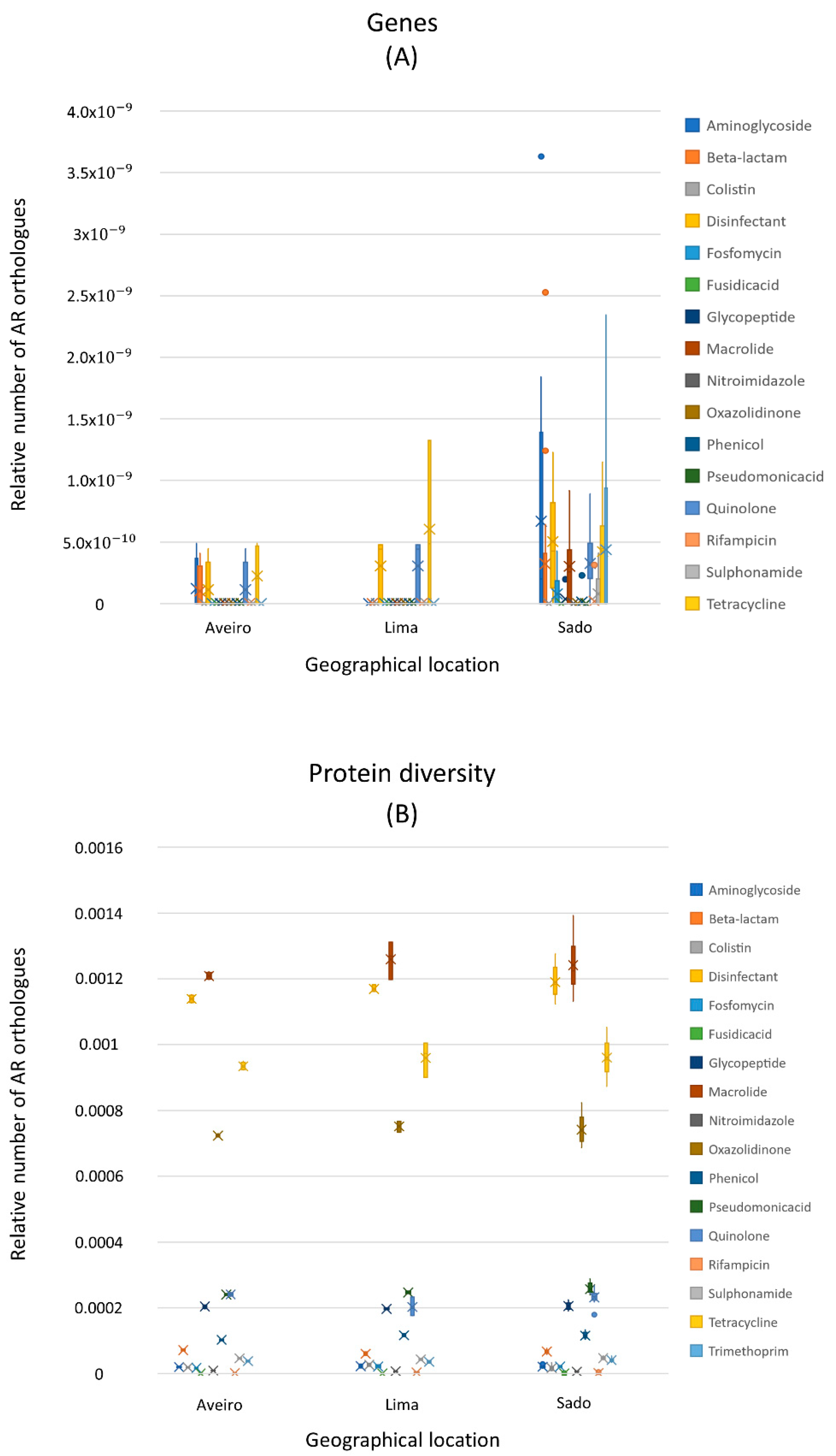
3.2. Antibiotic Resistance Encoding Genes
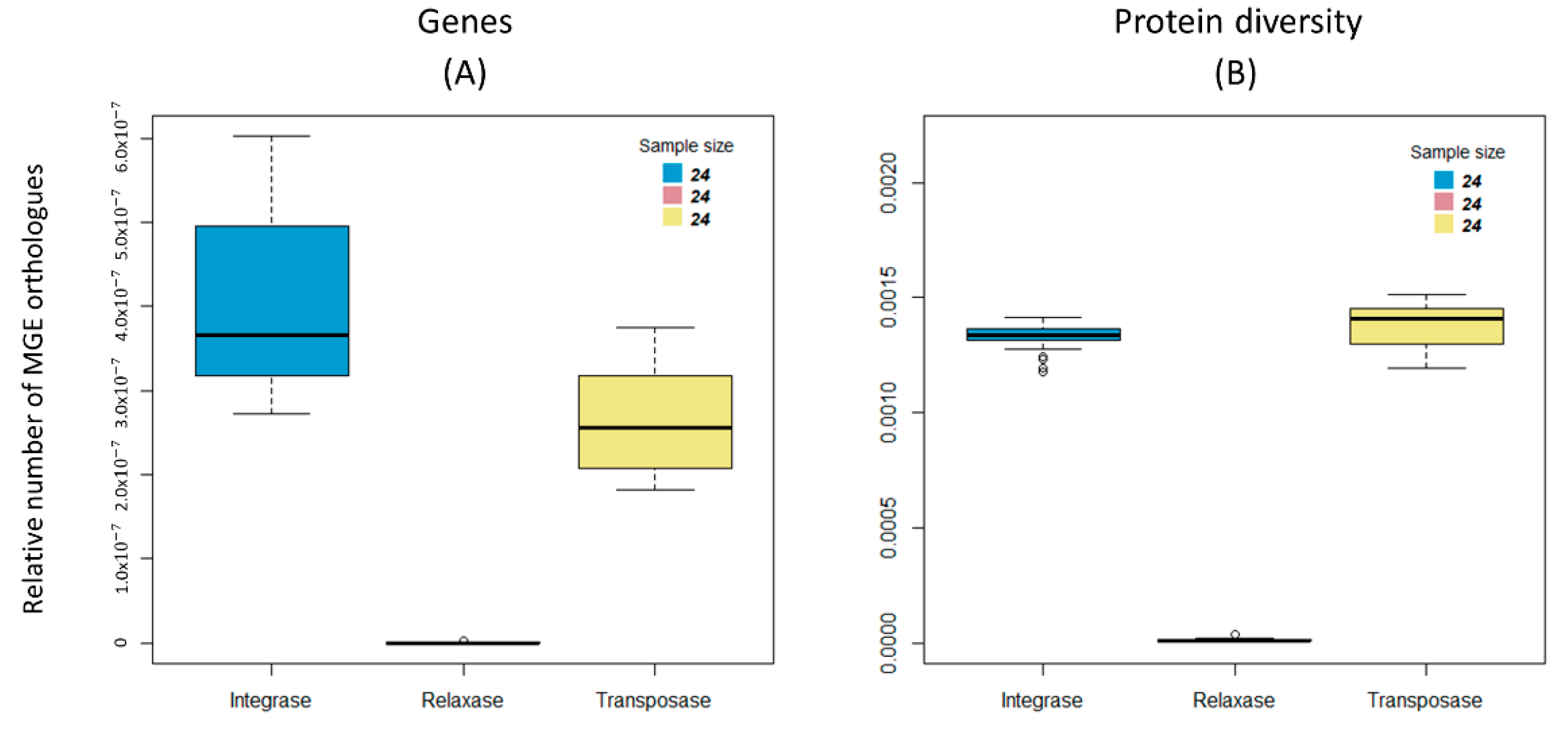
3.3. Distribution of the Mobile Genetic Elements
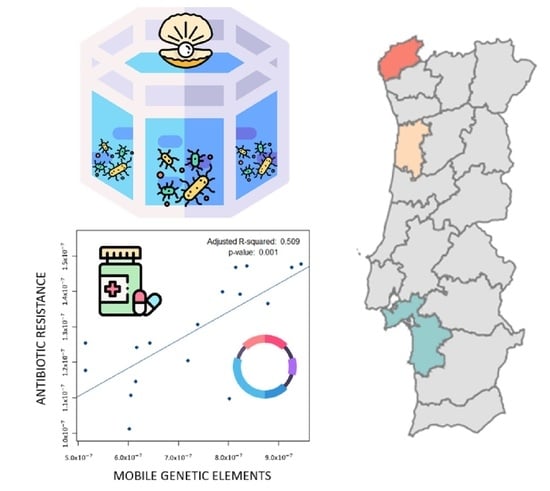
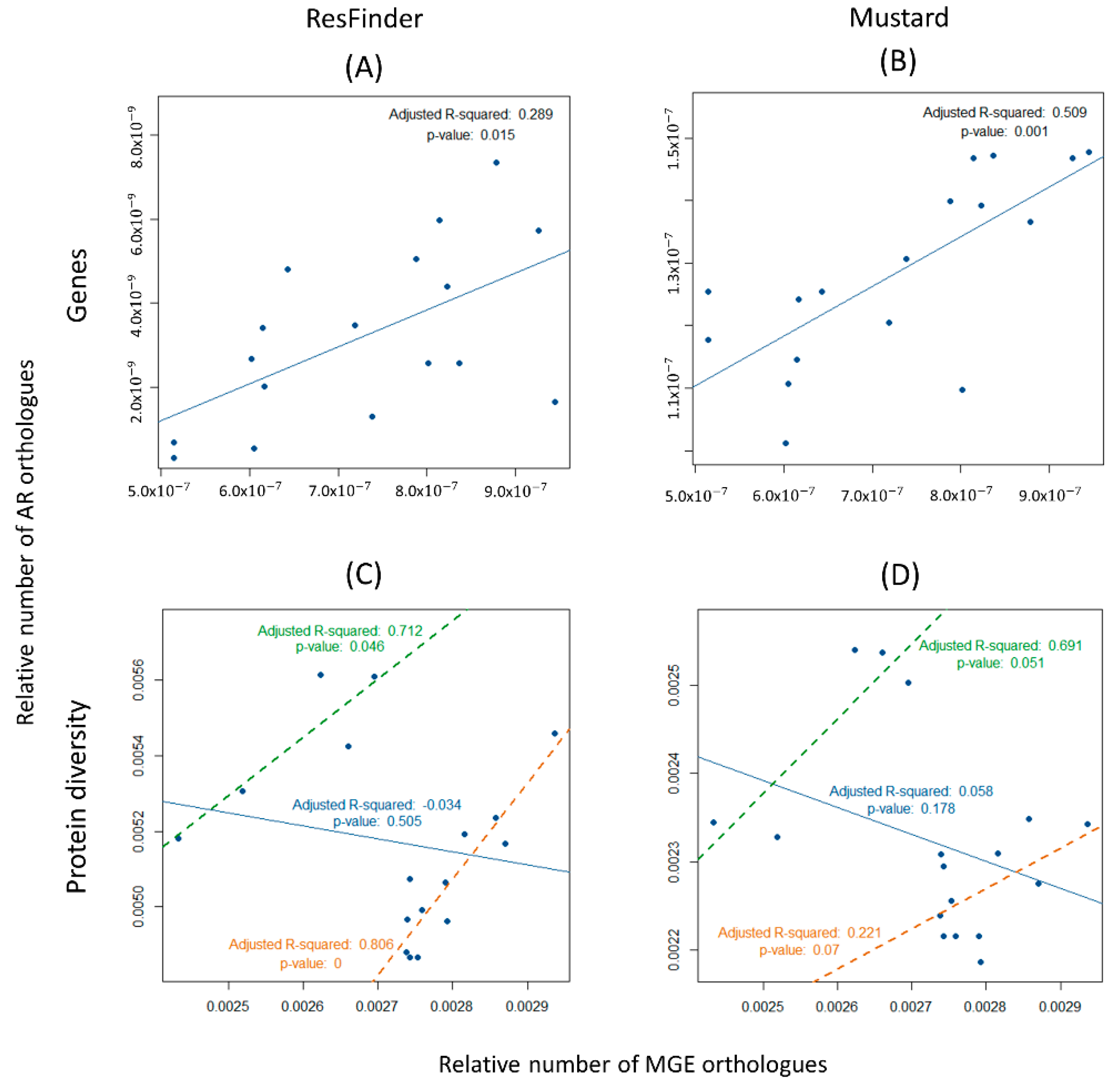
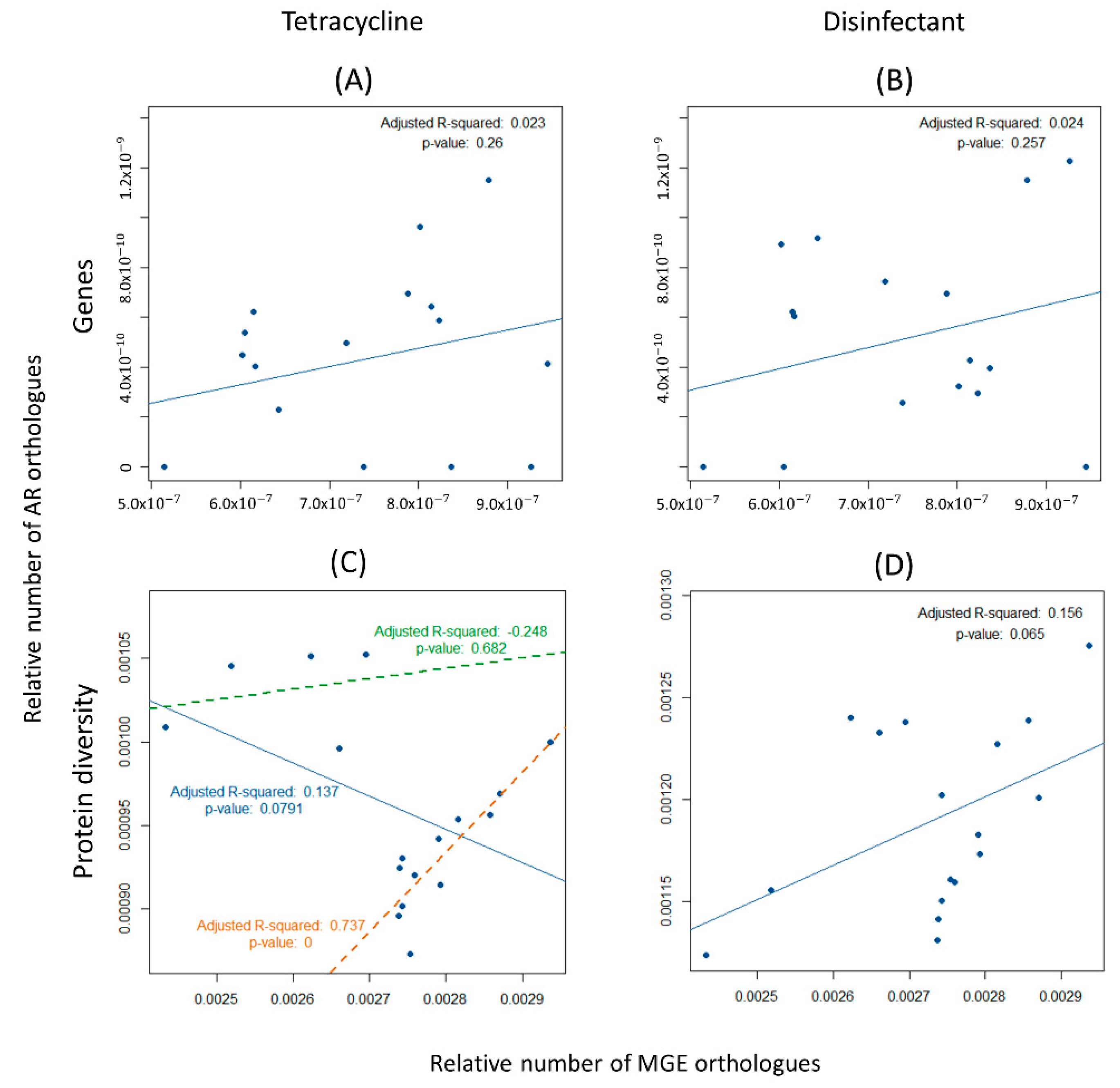
3.4. Spreading Potential of Antibiotic Resistance
4. Discussion
4.1. Resistome Fingerprint of Sediments of Estuarine Aquacultures
4.2. Spreading Potential and Dissemination of ARs in Aquaculture Environments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO Fisheries & Aquaculture. Available online: https://www.fao.org/fishery/en/facp (accessed on 26 September 2022).
- Revistia. ICSS XVI Paris, Proceedings Book. In Proceedings of the 16th International Conference on Social Sciences, Paris, France, 23–24 November 2018. [Google Scholar]
- FEAP. Federation of European Aquaculture Producers (FEAP) European AquacultureProduction Report 2014-2020. Available online: https://feap.info/wp-content/uploads/2022/03/production-report-v1.1.pdf (accessed on 16 November 2022).
- Rocha, C.P.; Cabral, H.N.; Marques, J.C.; Gonçalves, A.M.M. A Global Overview of Aquaculture Food Production with a Focus on the Activity’s Development in Transitional Systems—The Case Study of a South European Country (Portugal). J. Mar. Sci. Eng. 2022, 10, 417. [Google Scholar] [CrossRef]
- Korzekwa, K.; Gomułka, P.; Nowosad, J.; Kucharczyk, D.J.; Targońska, K.; Czarkowski, T.K.; Żarski, D.; Horváth, L.; Müller, T.; Kujawa, R.; et al. Bacterial Survey of Skin Ulcers in Anguilla Anguilla L. Females during Artificial Maturation under Controlled Conditions. Aquac. Rep. 2022, 25, 101259. [Google Scholar] [CrossRef]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the Wild: Antibiotic Resistance Genes in Natural Environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Done, H.Y.; Venkatesan, A.K.; Halden, R.U. Does the Recent Growth of Aquaculture Create Antibiotic Resistance Threats Different from Those Associated with Land Animal Production in Agriculture? AAPS J. 2015, 17, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, T.; Botelho, A. Metagenomics and Other Omics Approaches to Bacterial Communities and Antimicrobial Resistance Assessment in Aquacultures. Antibiotics 2021, 10, 787. [Google Scholar] [CrossRef]
- Santos, L.; Ramos, F. Antimicrobial Resistance in Aquaculture: Current Knowledge and Alternatives to Tackle the Problem. Int. J. Antimicrob. Agents 2018, 52, 135–143. [Google Scholar] [CrossRef]
- Reddy, S.; Kaur, K.; Barathe, P.; Shriram, V.; Govarthanan, M.; Kumar, V. Antimicrobial Resistance in Urban River Ecosystems. Microbiol. Res. 2022, 263, 127135. [Google Scholar] [CrossRef]
- Xu, M.; Huang, X.; Shen, X.; Chen, H.; Li, C.; Jin, G.; Cao, J.; Xue, Z. Metagenomic Insights into the Spatiotemporal Responses of Antibiotic Resistance Genes and Microbial Communities in Aquaculture Sediments. Chemosphere 2022, 307, 135596. [Google Scholar] [CrossRef]
- Port, J.A.; Cullen, A.C.; Wallace, J.C.; Smith, M.N.; Faustman, E.M. Metagenomic Frameworks for Monitoring Antibiotic Resistance in Aquatic Environments. Environ. Health Perspect. 2014, 122, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Bai, X.; Jing, L.; Chen, R.; Teng, Y. Characterization of Antibiotic Resistance Genes in the Sediments of an Urban River Revealed by Comparative Metagenomics Analysis. Sci. Total Environ. 2019, 653, 1513–1521. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.-X.; Huang, K.; Miao, Y.; Shi, P.; Liu, B.; Long, C.; Li, A. Metagenomic Profiling of Antibiotic Resistance Genes and Mobile Genetic Elements in a Tannery Wastewater Treatment Plant. PLoS ONE 2013, 8, e76079. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Sun, Y.; Zhang, T.; Ding, X.; Li, Y.; Wang, M.; Zeng, Z. Antibiotics, Antibiotic Resistance Genes, and Bacterial Community Composition in Fresh Water Aquaculture Environment in China. Microb. Ecol. 2015, 70, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Reverter, M.; Sarter, S.; Caruso, D.; Avarre, J.-C.; Combe, M.; Pepey, E.; Pouyaud, L.; Vega-Heredía, S.; de Verdal, H.; Gozlan, R.E. Aquaculture at the Crossroads of Global Warming and Antimicrobial Resistance. Nat. Commun. 2020, 11, 1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurya, A.P.; Rajkumari, J.; Pandey, P. Enrichment of Antibiotic Resistance Genes (ARGs) in Polyaromatic Hydrocarbon–Contaminated Soils: A Major Challenge for Environmental Health. Environ. Sci. Pollut. Res. 2021, 28, 12178–12189. [Google Scholar] [CrossRef]
- Wu, N.; Liang, J.; Wang, X.; Xie, S.; Xu, X. Copper Stimulates the Incidence of Antibiotic Resistance, Metal Resistance and Potential Pathogens in the Gut of Black Soldier Fly Larvae. J. Environ. Sci. 2021, 107, 150–159. [Google Scholar] [CrossRef]
- Brito, A.C.; Pereira, H.; Picado, A.; Cruz, J.; Cereja, R.; Biguino, B.; Chainho, P.; Nascimento, Â.; Carvalho, F.; Cabral, S.; et al. Increased Oyster Aquaculture in the Sado Estuary (Portugal): How to Ensure Ecosystem Sustainability? Sci. Total Environ. 2023, 855, 158898. [Google Scholar] [CrossRef]
- Silva, I.; Tacão, M.; Henriques, I. Selection of Antibiotic Resistance by Metals in a Riverine Bacterial Community. Chemosphere 2021, 263, 127936. [Google Scholar] [CrossRef]
- Nogueira, T.; Silva, D.G.; Lopes, S.; Botelho, A. Database of Metagenomes of Sediments from Estuarine Aquaculture Farms in Portugal—AquaRAM Project Collection. 2022. [Google Scholar]
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A Fast and Accurate Adapter Trimmer for next-Generation Sequencing Paired-End Reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 14 November 2022).
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The Metagenomics RAST Server—A Public Resource for the Automatic Phylogenetic and Functional Analysis of Metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Ruppé, E.; Ghozlane, A.; Tap, J.; Pons, N.; Alvarez, A.-S.; Maziers, N.; Cuesta, T.; Hernando-Amado, S.; Clares, I.; Martínez, J.L.; et al. Prediction of the Intestinal Resistome by a Three-Dimensional Structure-Based Method. Nat. Microbiol. 2019, 4, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids Using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 27 May 2022).
- Ogle, D.; Doll, J.; Wheeler, P.; Briand, C. FishR-Core-Team/FSA; Zenodo 2022. Available online: https://zenodo.org/record/6098468#.Y3rYsX1BxPY (accessed on 16 November 2022).
- Rebelo, J.S.; Domingues, C.P.F.; Dionisio, F.; Gomes, M.C.; Botelho, A.; Nogueira, T. COVID-19 Lockdowns May Reduce Resistance Genes Diversity in the Human Microbiome and the Need for Antibiotics. Int. J. Mol. Sci. 2021, 22, 6891. [Google Scholar] [CrossRef] [PubMed]
- Nucleotide Sequence and Organization of the Multiresistance Plasmid PSCFS1 from Staphylococcus Sciuri | Journal of Antimicrobial Chemotherapy | Oxford Academic. Available online: https://academic.oup.com/jac/article/54/5/936/812075 (accessed on 27 September 2022).
- Escudeiro, P.; Pothier, J.; Dionisio, F.; Nogueira, T. Antibiotic Resistance Gene Diversity and Virulence Gene Diversity Are Correlated in Human Gut and Environmental Microbiomes. mSphere 2019, 4, e00135-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darmancier, H.; Domingues, C.P.F.; Rebelo, J.S.; Amaro, A.; Dionísio, F.; Pothier, J.; Serra, O.; Nogueira, T. Are Virulence and Antibiotic Resistance Genes Linked? A Comprehensive Analysis of Bacterial Chromosomes and Plasmids. Antibiotics 2022, 11, 706. [Google Scholar] [CrossRef]
- Rico, A.; Satapornvanit, K.; Haque, M.M.; Min, J.; Nguyen, P.T.; Telfer, T.C.; van den Brink, P.J. Use of Chemicals and Biological Products in Asian Aquaculture and Their Potential Environmental Risks: A Critical Review. Rev. Aquac. 2012, 4, 75–93. [Google Scholar] [CrossRef]
- De Briyne, N. Critically Important Antibiotics-Comparison Table WHO, OIE and AMEG. Available online: https://www.researchgate.net/publication/328981153_Critically_Important_Antibiotics-_comparison_table_WHO_OIE_and_AMEG (accessed on 16 January 2020).
- Marano, R.B.M.; Fernandes, T.; Manaia, C.M.; Nunes, O.; Morrison, D.; Berendonk, T.U.; Kreuzinger, N.; Tenson, T.; Corno, G.; Fatta-Kassinos, D.; et al. A Global Multinational Survey of Cefotaxime-Resistant Coliforms in Urban Wastewater Treatment Plants. Environ. Int. 2020, 144, 106035. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, Y.; Henderson, I.R.; Guo, J. Artificial Sweeteners Stimulate Horizontal Transfer of Extracellular Antibiotic Resistance Genes through Natural Transformation. ISME J. 2022, 16, 543–554. [Google Scholar] [CrossRef]
- Singh, N.S.; Singhal, N.; Kumar, M.; Virdi, J.S. High Prevalence of Drug Resistance and Class 1 Integrons in Escherichia Coli Isolated From River Yamuna, India: A Serious Public Health Risk. Front. Microbiol. 2021, 12, 621564. [Google Scholar] [CrossRef]
- Mafuna, T.; Matle, I.; Magwedere, K.; Pierneef, R.E.; Reva, O.N. Comparative Genomics of Listeria Species Recovered from Meat and Food Processing Facilities. Microbiol. Spectr. 2022, 10, e01189-22. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kruskal–Wallis (p-Value) | Dunn’s Test (Adjusted p-Value) | ||
|---|---|---|---|
| Resfinder (Protein Diversity) | 0.543 | Aveiro–Lima | 0.709 |
| Aveiro–Sado | 0.905 | ||
| Lima–Sado | 0.832 | ||
| Resfinder (Genes) | 0.034 | Aveiro–Lima | 0.677 |
| Aveiro–Sado | 0.060 | ||
| Lima–Sado | 0.240 | ||
| Mustard (Protein diversity) | 0.087 | Aveiro–Lima | 0.412 |
| Aveiro–Sado | 0.082 | ||
| Lima–Sado | 0.677 | ||
| Mustard (Genes) | 0.001 | Aveiro–Lima | 0.829 |
| Aveiro–Sado | 0.004 | ||
| Lima–Sado | 0.021 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, D.G.; Domingues, C.P.F.; Figueiredo, J.F.; Dionisio, F.; Botelho, A.; Nogueira, T. Estuarine Aquacultures at the Crossroads of Animal Production and Antibacterial Resistance: A Metagenomic Approach to the Resistome. Biology 2022, 11, 1681. https://doi.org/10.3390/biology11111681
Silva DG, Domingues CPF, Figueiredo JF, Dionisio F, Botelho A, Nogueira T. Estuarine Aquacultures at the Crossroads of Animal Production and Antibacterial Resistance: A Metagenomic Approach to the Resistome. Biology. 2022; 11(11):1681. https://doi.org/10.3390/biology11111681
Chicago/Turabian StyleSilva, Daniel G., Célia P. F. Domingues, João F. Figueiredo, Francisco Dionisio, Ana Botelho, and Teresa Nogueira. 2022. "Estuarine Aquacultures at the Crossroads of Animal Production and Antibacterial Resistance: A Metagenomic Approach to the Resistome" Biology 11, no. 11: 1681. https://doi.org/10.3390/biology11111681